Information of protein 3-dimensional (3D) structures plays an essential role in molecular biology, cell biology, biomedicine, and drug design. Accordingly, predicting the 3D structure of a protein is an important problem in Bioinformatics.
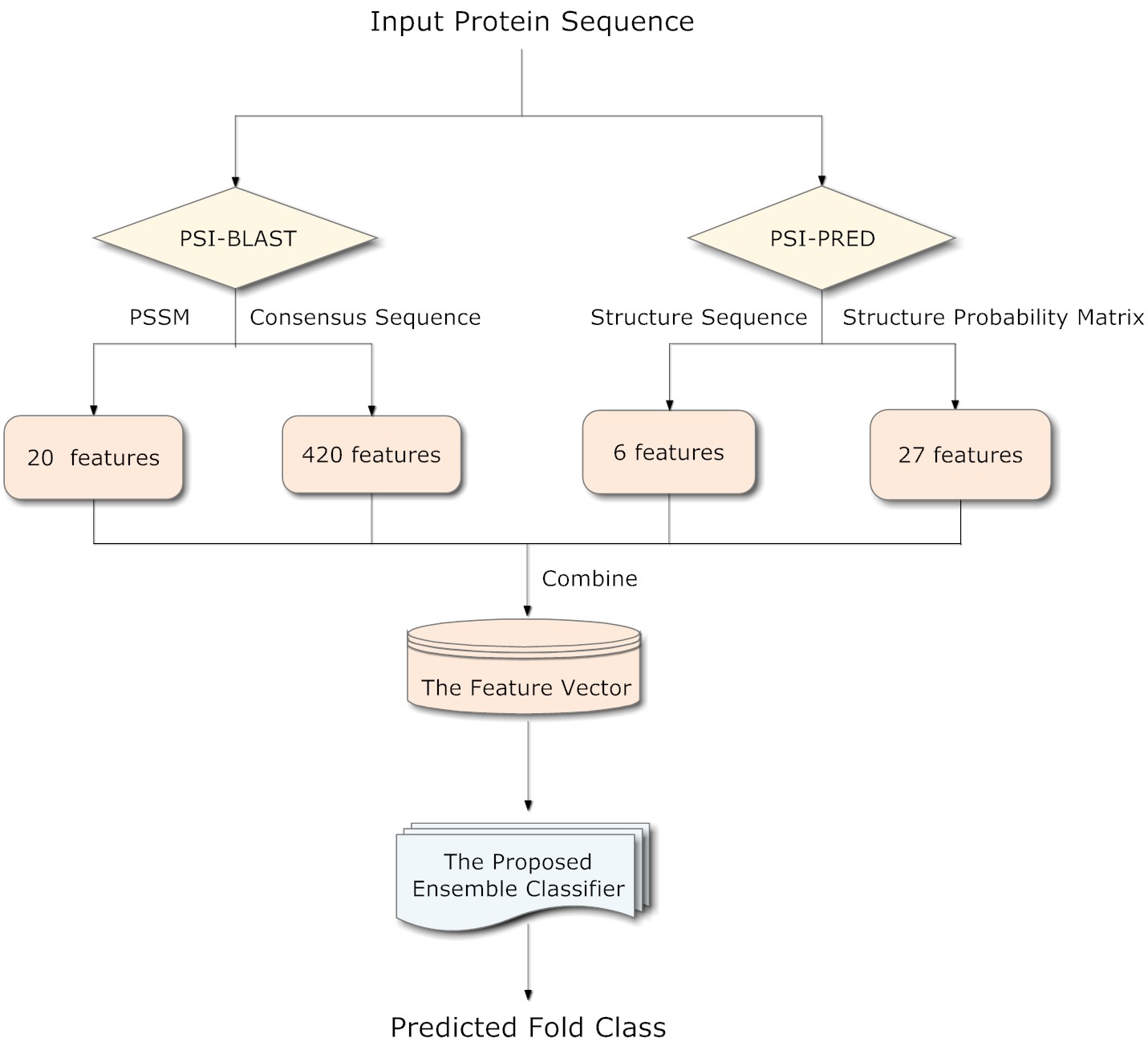
In this paper, we propose a novel taxonomic method, called PFPA. It is featured by combining a comprehensive feature set through an ensemble classifier. The proposed feature set sufficiently explores the discriminatory sequence evolution information from the profile of PSI-BLAST, and the local and global secondary structure information from the profile of PSI-PRED. As compared with existing taxonomic methods on a benchmark dataset (called DD), PFPA successfully improves the prediction accuracy to 73.6%, which is 2.1% higher than the best accuracy ever reported. The significant improvement suggests the superiority of PFPA. Moreover, PFPA performs well without significant performance degradation on three updated large-scale datasets, which suggests the robustness and generalization of PFPA.
More details of PFPA can be seen in the paper. The procedure of how PFPA works can be seen below.